

Contemporary diagnostic and therapeutic approaches to ATTR cardiac amyloidosis
Written by Dr Carlos N. Perez-Garcia, Advanced Heart Failure and Cardiac transplant fellow and Professor Emer Joyce, Heart Function and Transplant Cardiologist – Mater Misericordiae University Hospital, Dublin, Ireland – Clinical Professor, UCD School of Medicine


Introduction
The term ‘amyloidosis ’ describes a group of disorders characterized by abnormal protein folding, leading to the aggregation and accumulation of these misfolded proteins, or ‘amyloid fibrils’ in the tissues of affected organs. These amyloid fibrils display the pathognomonic property of apple-green birefringence when viewed under cross polarized light after staining with Congo red. 1 The deposit of these insoluble proteins into tissues leads to marked interstitial expansion, direct cell toxicity and cell death.
In case of heart involvement, amyloidosis is associated with a particularly high morbidity and mortality. 2.3 Although there are more than 30 known amyloidogenic proteins, only 9 accumulate in the myocardium, and the two most frequent etiologies of cardiac amyloidosis (CA) are misfolding of either the transthyretin tetramer (ATTR) or immunoglobulin light-chains secreted by clonal plasma cells (AL), which combined account for 95% of cases of CA. 4
Although the focus of this article is centered on the diagnostic and therapeutic advances in ATTR-CA, it is critical that a diagnosis of AL amyloidosis is appropriately ruled out in all patients suspected of having cardiac amyloidosis.
ATTR-CA is being increasingly recognized by the general medical and cardiology community due to a combination of greater awareness of the clinical syndrome and associated cardiac imaging findings alongside recent emergence of a validated noninvasive pathway for its diagnosis.
Patients can be classified in two groups: either hereditary (hATTR- CA) or wild type (wtATTR-CA), depending on the presence or absence, respectively, of a mutation in the TTR gene. 5 More than 120 mutations have been reported in the TTR gene, located on chromosome 18, and, despite the fact that it is an autosomaldominant disorder, its penetrance is incomplete and considerable phenotypic and geographical heterogeneity is characteristic.
Clinical syndromes can vary extensively from a neurologicalpredominant phenotype to a cardiac-predominant phenotype or a spectrum of both. 6 The Thr60Ala or ‘T60A’ variant is the dominant variant in Ireland and in the landmark study by Reilly et al. in 1995, was found to affect up to 1% of the population of North Donegal over the age of 45, with an estimated penetrance of 80%. This variant causes both neurologic and cardiac phenotypes, with typical onset of symptoms in the 5th and 6th decades. 7
The true prevalence of wtATTR- CA is likely to be significantly higher than previously described, possibly occurring in 1-3% of those >75 years of age, increasing to 14% of those >85 years. 8
Recent studies also estimate that wtATTR accounts for 13% of elderly patients hospitalized with heart failure (HF) with preserved ejection fraction and 16% of patients undergoing transcatheter aortic valve replacement. 9,10 It is not yet clear exactly why the normal genetic sequence TTR protein becomes unstable and aggregates, but it is thought likely related to aging.
Current diagnostic strategies for ATTR-CA
Traditionally CA has been an underdiagnosed condition, primarily due to lack of recognition compounded by non-specific symptoms and the presence of co-morbidities in this age cohort. Delayed diagnosis is common, with an average lead-in time from initial symptoms to definitive diagnosis of at least 6 months. 11,12 A recent European study revealed that 35% of a wtATTR-CA cohort had been previously misdiagnosed with other cardiovascular diseases commonly recognized in clinical practice, including hypertensive cardiomyopathy (35%), hypertrophic cardiomyopathy (23.5%), ischaemic heart disease (11.8%), and aortic stenosis (8.8%). 13
Recent advances in cardiac imaging, diagnostic strategies, and therapies have enabled earlier recognition and treatment of CA, which is critical due to the fact that CA is progressive and life-threatening if left untreated. Currently, the diagnosis of CA can be divided in two phases: an initial screening phase and a definitive diagnosis phase1, and it requires a comprehensive approach considering clinical red flags, biomarkers, ECG, multimodality imaging and, in certain cases, may still require invasive tissue biopsy including native heart biopsy.
1. Screening of cardiac amyloidosis. When to suspect it?
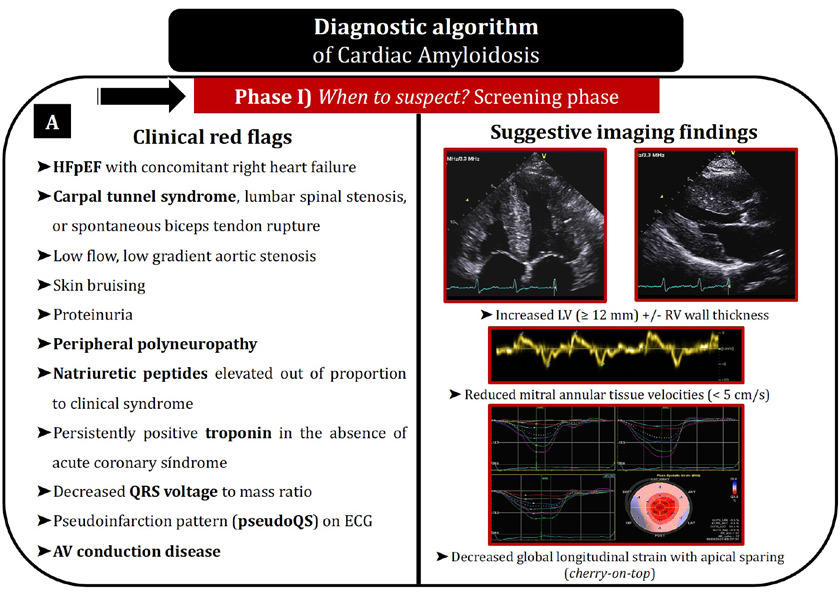
From a clinical perspective, CA typically appears associated with several cardiac and extracardiac manifestations which are variable depending on the type of CA and can be useful to suspect the disease. These signs and symptoms are known as red flags and are shown in Figure 1A. At a cardiac level, the presence of HF with an unexplained increased left and right ventricular wall thickness, disproportionally elevated natriuretic peptides, persistently positive troponin in the absence of acute coronary syndrome, low QRS voltages or early conduction system disease are also findings that could evoke CA. Other clinical clues to take into consideration are the presence of low-flow, low-gradient aortic stenosis, the need for downtitration or discontinuation of antihypertensive therapies, as well as the intolerance of beta-blockade in newly diagnosed HF.1,9
 • 12- lead electrocardiogram (ECG)
• 12- lead electrocardiogram (ECG)
An abnormal ECG is present in >90% of patients with CA. As mentioned above, discordant low QRS voltages in the limb leads in spite of left ventricle increased wall thickness is suggestive of CA, and its presence portends a worse outcome in CA patients. 14 A pseudo infarction pattern, defined as QS waves in any two consecutive leads, is also a typical finding described in CA. Interestingly, these two findings may be present irrespective of increased ventricular wall thickness and both of them are more frequent in AL than in ATTR-CA (only 25% to 40% of patients with ATTR-CA meet low-voltage criteria), hence their absence does not exclude the possibility of CA. 15,16 Apart from that, as previously mentioned, the presence of conduction system disease or atrial fibrillation are significantly frequent.
• Echocardiography
Echocardiography is an essential tool in the screening of CA, typically revealing increased left and right ventricular wall thickness with a characteristic speckled appearance, valvular thickening, bi-atrial enlargement, reduced mitral annular tissue velocities (<5 cm/s), and not rarely, pericardial effusion. 1, 5 In terms of cardiac deformation, the decreased global longitudinal LV strain (absolute value < -15%) is suggestive of CA, and there is a distinctive pattern of apical sparing in which the left ventricular apical region shows more normal strain compared with progressively worse values at the mid and basal regions (‘cherry-on-top’ pattern on the bull’s eye plot of global longitudinal strain, Figure 1A). This apical sparing pattern was shown to have 93% sensitivity and 82% specificity in the diagnosis of cardiac amyloidosis. 18
• Cardiac magnetic resonance
Because of its tissue characterization capabilities, cardiac magnetic resonance (CMR) is a versatile imaging technique to assess CA by late gadolinium enhancement (LGE) sequences and extracellular volume (ECV) mapping. In CA, the pattern of LGE is heterogenous. It typically begins as subendocardial and progresses to transmural involvement in the later stages of disease. Furthermore, gadolinium kinetics are usually abnormal, characterized by a suboptimal myocardial nulling. 16,19 Several investigations have shown that the presence of LGE on CMR is a robust predictor of outcome, independent of other known cardiac biomarkers. 20
The use of pre-and post-gadolinium T1 mapping allows for the quantification of ECV, which is the percentage of myocardium composed of the extracellular space. ECV is markedly elevated in CA, being strongly supportive of the diagnosis when it is ≥0.40%, and correlating with histologic amyloid burden. 1, 21 The degree of myocardial edema, measured with the use of T2 imaging, has also been shown to be an independent prognostic marker. 22
2. Definitive diagnosis phase. Diagnostic algorithm.
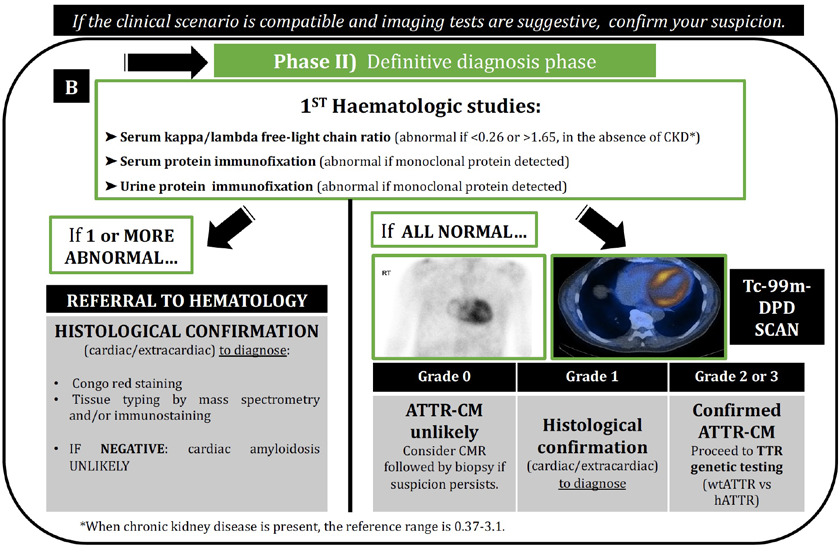
When CA is suspected on the basis of clinical or imaging findings, a timely, definite diagnosis must be obtained in order to avoid delayed treatments and disease progression. The current algorithm (Figure 1B) requires that all the patients with suspected CA must undergo screening for monoclonal proteins by quantification of serum free light chains together with serum and urine immunofixation, in order to identify the potential need for further hematological and/ or histological investigation to exclude AL-CA.
As illustrated in our algorithm, this should always be the first step following clinical suspicion of CA. If there is no monoclonal protein detected and free-light chain ratio is normal, the second step will be a cardiac scintigraphy with a bone-avid radiotracer (99mTc-PYP, DPD or HMDP), which can confirm the diagnosis of ATTR-CA via a non-invasive diagnosis pathway. 1, 17, 23 However, if 1 or more of these initial 3 hematological tests are abnormal, the referral for Hematology investigation and /or cardiac or extracardiac biopsy of involved.
• Cardiac scintigraphy
The ability of myocardial scintigraphy (typically in Ireland using Tc-99m- 3,3-diphosphono-1,2 propanodicarboxylic acid [DPD)]) to provide a non-invasive diagnostic pathway for ATTR-CA, if certain criteria are met, is one of the principal game changers that has emerged for this condition over recent years. The theoretical basis of why these tracers bind to TTR is still not well understood, but has been attributed to the presence of microcalcifications that are more common in ATTR than AL cardiac tissue. Therefore, it is more specific to ATTR-CA, although scintigraphy can return a positive result in 7-9% of AL- CA cases.
Other rare causes of potential false positive results like hydroxychloroquine-induced restrictive cardiomyopathy have been reported. 25, 26 Interestingly, quantification of tracer retention in the myocardium reflects the severity of myocardial involvement, and it is assessed by a semiquantitative scheme (Perugini score), which compares cardiac tracer uptake with bone uptake of the rib, grading from 0 (no uptake) to 3 (cardiac uptake exceeds rib).27
When grade ≥2 uptake is present in the absence of a serum or urine monoclonal light chain protein, it has been demonstrated that a reliable diagnosis of ATTR- CM can be made non-invasively with 99% of specificity. 28 This diagnostic pathway has greatly increased our ability to diagnose ATTR-CA and has consequently improved patient access to potentially life-prolonging ATTR therapies.
 • Endomyocardial and other tissue biopsy
• Endomyocardial and other tissue biopsy
Endomyocardial biopsy (EMB) remains the gold standard for CA diagnosis and is nearly 100% sensitive and specific if biopsy specimens are collected from multiple sites (≥4 are recommended) and tested for amyloid deposits by Congo red staining. 29
Identification of amyloid must be followed by classification of the amyloid fibril protein, so amyloid subtype is then determined with either laser dissection with mass spectroscopy, immunohistochemistry or immunoelectron microscopy in specialized centres. 30 Compared to EMB, other tissue biopsies, such as gastrointestinal or abdominal fat aspirate, have remarkably varying sensitivity (for example, only 15% for fat aspirate in wtATTR-CA patients). 5
As shown in our algorithm, it is important to remember that invasive tissue biopsy is always required if AL-CA is suspected; however, in ATTR-CA, invasive biopsy is generally reserved when the initial non-invasive testing is inconclusive. 1, 23 In addition, it is important to remember that up to 40% of patients with ATTR-CA can have a monoclonal gammopathy of unknown significance (MGUS) and scintigraphy alone cannot ensure a diagnosis with 100% specificity. In this setting, endomyocardial biopsy is necessary to have a definitive diagnosis of ATTR-CA. 23
• Genetic testing
All patients diagnosed with ATTR-CA should undergo genetic testing to screen for a pathogenic gene mutation. In the case of Ireland, Thr60Ala (T60A) is the most prevalent 7 , and the genetic testing has implications, not only for the patient, but also for family members, since it facilitates an earlier diagnosis and, in case of hATTR-CA, better outcomes if treated at an earlier stage of the disease. Currently, the ideal method and intervals for organ screening for hATTR carriers remain unknown.
Current and future therapies for ATTR-CA
Prompt diagnosis of the CA subtype is critical to guide specific treatment, as therapy is more effective in the earlier stages of the disease. This premise is important in all types of CA, but especially in AL in which, whenever possible, patients must be transferred to specialized centers and specific chemotherapy regimens must be undertaken by multidisciplinary teams, involving oncohematology and cardiology specialists.
Therapies for patients with ATTR-CA can be classified in two groups: those which treat complications and comorbidities (including treatment of heart failure, arrhythmias, conduction disturbances, thromboembolism, concomitant presence of severe aortic stenosis…), and those which are targeted, diseasemodifying therapies. In this section, the focus is on the latter, the emergence of which in recent years represent a watershed in the management and outcomes of patients with ATTR-CM.
In 2018, 3 large clinical trials transformed the landscape of ATTR amyloidosis treatment. 31-33 On the basis of these results, the US Food and Drug Administration (FDA) approved tafamidis for the treatment of ATTR-CA (regardless of type) and both patisiran and inotersen for hATTR polyneuropathy. Considering their mechanism of action, ATTR therapies can be divided into three main categories, depending if they 1) stabilize circulating TTR molecules (stabilizers), 2) suppress TTR synthesis (liver transplantation, genetic silencers and gene editing), or 3) promote the disruption and/or extraction of the deposits. Table 1 summarizes the current available therapies for ATTR-CA.
Transthyretin stabilizing molecule therapies
-Tafamidis is a daily oral medication, which binds the thyroxine-binding sites of TTR with high affinity and selectivity, slowing dissociation of TTR tetramers into monomers, which consequently inhibits aggregation. Interestingly, Tafamidis was inspired by the discovery of a stabilizing mutation in transthyretin (Thr119Met), which protects Val30Met mutation carriers from developing amyloidosis. 34

Initially it was approved for the treatment of stage 1 and 2 hATTR polyneuropathy 35 , and it was not until the results of the phase 3 ATTR-ACT randomized clinical trial that was approved for patients with ATTR-CA, either wild type or hereditary. In this study, tafamidis (n=264) was associated with reductions in all-cause mortality and cardiovascular-related hospitalizations and reduced the decline in functional capacity and quality of life as compared with placebo (n=177).
Although improvements in biomarkers were noted starting at 9 months, it was not until after 18 months that a survival benefit was observed. 31 Notably, while the benefits were consistent in those with NYHA I-II symptoms, in those with NYHA class III symptoms, increased rates of hospitalizations were observed in the tafamidis-treated group.30
Based on these data, tafamidis has been approved for all patients with ATTR-CA and is indicated per the recently updated ESC 2021 HF guidelines. 36 At the time of writing, the drug has not yet been approved for reimbursement by the HSE, but this is expected to be updated shortly.
-Diflunisal is a generic nonsteroidal anti-inflammatory drug with a stabilizer mechanism. A randomized placebo-controlled trial demonstrated efficacy of diflunisal in reducing the progression of hATTR polyneuropathy 37 , and a retrospective study in patients with cardiomyopathy demonstrated some evidence of cardiac stabilization. 38 Nevertheless, there is no prospective data in ATTR- CA and diflunisal is one of those off-label therapies in ATTR-CA, whose use is at the discretion of the treating physician. Due to its nonsteroidal anti-inflammatory properties, gastroprotection and monitoring renal function are recommended.
-Investigational transthyretinstabilizing molecule therapies: an additional oral drug that stabilizes the transthyretin tetramer similar to tafamidis, acoramidis (AG-10), is currently being tested in patients with ATTR-CA in a large, phase 3 clinical trial (ATTRIBUTE-CM). 39
Inhibitory oligonucleotide therapies (genetic silencers)
-Patisiran is an oligo-nucleotidebased therapeutic agent that originally circulates as a double-stranded RNA molecule, subsequently dissociating into single-stranded molecules in the cytoplasm. These strands functions as a small interfering RNA (siRNA), binding the transthyretin messenger RNA (mRNA) in the cytoplasm to form an RNA-induced silencing complex. This process leads to mRNA cleavage, inhibiting the synthesis of the TTR protein and thereby decreasing levels of both normal and mutant forms.
The effects of patisiran in hATTR neuropathy were studied in the APOLLO trial, where patisirantreated patients had significant improvement in neuropathy scores and gait speed compared to baseline. 32 In a subgroup analysis of patients with ATTR-CA, patisiran was associated with benefit over placebo in NT-proBNP, left ventricular wall thickness, and longitudinal strain measures. 40 Following these results, patisiran has been approved for patients with hATTR polyneuropathy with or without cardiac involvement, but not for those without polyneuropathy.
It is administered intravenously every 3 weeks and patients need to be premedicated with a corticosteroid, paracetamol and antihistamines to reduce the risk of an infusion reaction. After receiving the drug, patients must be monitored for symptoms which can include flushing, back pain, nausea, abdominal pain, shortness of breath, or headache. Currently in Ireland, both hospital and home infusions have recently emerged as possible options for approved patients through a managed medicines program. Due to the fact that transthyretin aids in vitamin K transport via a complex with vitamin A, all patients treated should be prescribed vitamin A supplementation to avoid deficiencies and, subsequently, ocular toxicity. 17
-Inotersen is an antisense oligonucleotide which binds to the target mRNA in the nucleus, promoting its cleavage. Administered as a weekly subcutaneous injection, Inotersen is associated with greater stabilization in neuropathy scores in hATTR neuropathy compared to placebo, and in a substudy of patients with ATTR-CA, inotersen demonstrated stabilization of disease as measured by left ventricular wall thickness, left ventricular mass, 6-min walk test, and echocardiographic global systolic strain. 33 In terms of adverse events, a higher risk of glomerulonephritis and thrombocytopenia were observed, and subsequently, a recent trial of inotersen in patients with ATTR-CA is currently on hold because of these findings. 41 It is not currently available for reimbursement in Ireland.
Elimination of deposits
Other off-label therapies in ATTR-CA include:
– Doxycycline and Ursodiol: shown to disaggregate formed amyloid fibrils in in vitro studies. 45 A retrospective study of 53 patients with ATTR-CA treated with both drugs showed no significant cardiac disease progression over 22 months 46 , and larger randomized clinical trials are currently under way to assess the safety and efficacy of this combination in ATTR and ATTR-CA. 47
– Epigallocatechin 3-gallate, most abundant catechin in green tea, has demonstrated disruption of transthyretin amyloid fibrils in mice models. 49 One observational study of 25 patients with wtATTR- CA treated with 600 mg of epigallocatechin 3-gallate daily for 1 year showed a decrease in left ventricular mass without change in left ventricular wall thickness by CMR. 50
Future therapeutic perspectives in transthyretin amyloidosis
Current therapeutic options for ATTR amyloidosis rely on reducing ongoing amyloid formation through stabilization of the tetrameric form of TTR or through inhibition of TTR protein synthesis by means of degradation of TTR mRNA. Such treatments produce symptom relief and functional improvement and prolong survival, but are limited by the requirement for long-term administration to maintain TTR knockdown. Furthermore, in the case of patients on stabilizers, disease progression persists. 51 In this setting, a promising alternative are the in vivo gene-editing therapies via Cas9 endonuclease (CRISPR-Cas9), to achieve a sustained knockdown of the TTR gene.
This is the case of NTLA-2001, a lipid nanoparticle with liver tropism that carries a single guide RNA (sgRNA), whose results in patients with hATTR in a phase 1, open-label, multicenter study are really optimistic. A single intravenous administration of NTLA-2001 led to a mean reduction of 52% and 87% in serum TTR protein concentrations in the groups that received a dose of 0.1 mg/ kg and 0.3 mg/kg, respectively, with no serious adverse events reported. 52 These results illustrate how CRISPR-Cas9–based in vivo gene-editing therapies provide a greater TTR knockdown than currently available therapies, and represent a paradigm shift in the management of this disease.
The Irish Experience
As discussed above, Irish ATTR patients, based on the prevalence of the T60A mutation, through both the National and diaspora experience, have played an important role in furthering knowledge and clinical insights into this previously underrecognized and poorly understood disease. The increased recognition and diagnosis of wtATTR-CA in Ireland also parallels international figures. Despite these facts, the current model of care for amyloidosis in Ireland still involves patients travelling to the National Amyloidosis Centre (NAC) in London.
A National Working Group for Amyloidosis was established by the HSE in 2019 to create a new model of care in this country for patients with this disease, chaired by Dr Mark Coyne. The Working Group Report was submitted in 2020 and implementation of its findings are awaited. There is no doubt that the COVID-19 pandemic imparted further stress on patients required to travel to continue to access optimal amyloidosis care.
It is hoped that that engagement of the HSE on this report, and its recommendations, will be forthcoming, particularly in light of the advent of reimbursement of disease-specific therapies as above, which although highly welcomed, nonetheless will highlight the extreme lack of any targeted funding to support the program in Ireland – including but not limited to timely access to required testing, necessary hiring of clinical staff to support clinical coordination (across sites as well as across hospital and home) and audit of newer therapies, as well as facilities for infusions, monitoring, clinical trial provision etc.
Conclusions
Cardiac amyloidosis, and specifically, ATTR-CA affects a growing population of patients encountered in our clinical practice. With the advent of contemporary non-invasive imaging techniques, diagnostics in this field have substantially improved, allowing an earlier detection of affected individuals. This alongside the emergence of effective specific therapies for ATTR-CA is expected to translate into improved outcomes, making possible a promising future for amyloid patients. It is hoped that with dedicated funding and National support and coordination, Ireland can be a future leader in ATTR amyloidosis care and best practice.
References available on request.
If you would like to receive an HPN magazine every month, complete the form below to be added to the mailing list. Only those who meet the expected criteria for readership will be added to the list. SUBSCRIBE
Follow us: